Duchenne Muscular Dystrophy (DMD) is a devastating and ultimately fatal genetic disease that affects approximately 1 in every 5000 boys, making it one of the most common human genetic diseases. The disease results from mutations in the X-linked DMD gene that prevent expression of its functional protein product, dystrophin. A large (427 kD), structural protein with functional domains at the amino- and carboxy-termini, dystrophin localizes at the cytoplasmic face of the sarcolemma and links the intracellular cytoskeleton to the extracellular matrix via actin and proteins in the dystrophin-associated glycoprotein complex. Dystrophin helps muscle fibers maintain their integrity and resiliency during muscular contraction, and without it, myofibers undergo cycles of damage-induced degeneration and regeneration until the muscle’s regenerative capacity is exhausted. Death usually occurs when patients reach their mid-20s from breathing difficulties or associated cardiomyopathies. Therapies for treating DMD have included viral-mediated delivery of DMD “minigenes” encoding shortened, but functional dystrophin protein and antisense oligonucleotide-based strategies that interfere with dystrophin splicing to remove specific exons in which DMD-causing mutations reside. Both strategies, however, present challenges: the very large size of the dystrophin cDNA has made designing functional constructs small enough to fit in delivery vectors difficult, and the antisense oligo approach requires repeated injections to retain expression of functional protein and has other potential and unwanted side-effects. Additional strategies that can provide long-term remediation of DMD are still being sought.
In January 2016, no less than three studies published in the journal Science and contributed by collaborative teams led by researchers at Harvard University and the Massachusetts Institute of Technology (Tabebordbar M et al. 2016), the University of Texas Southwestern Medical Center (Long C et al. 2016) and Duke University (Nelson, CE et al. 2016) reported using CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR-associated protein 9) genome editing approaches to permanently correct the genetic mutation in the well-known Dmdmdx mouse model for DMD (C57BL/10ScSn-Dmdmdx/J – 001801). The technique partially restored not only functional dystrophin expression, but also dystrophic muscle function. The studies validate the CRISPR/Cas9 genome editing platform as a potential therapeutic approach for treating DMD and other genetic diseases in human patients postnatally.
A CRISPR/Cas9-mediated, exon-skipping strategy to correct the Dmdmdx mutation
CRISPR/Cas9 system can introduce either variable or directed mutations at precise locations within the genome. The technique involves designing specific “single guide RNAs” (sgRNAs) that recognize specific sequences in the genome that are adjacent protospacer adjacent motif (PAM) sites. When only an sgRNA and Cas9 are introduced into a cell, small deletions (indels) are introduced adjacent to the PAM site via double-stranded DNA breaks and non-homologous end joining (NHEJ). When an extra DNA template is also supplied, however, precise genomic modifications, or knock-in constructs (e.g. loxP sites, EGFP cassettes, etc.) can be introduced via homology-directed DNA repair (for an overview see our 2014 blog post). Previously, the UT Southwestern team (Long C et al. 2016) had used CRISPR/Cas9 to correct the Dmdmdxmutation in the germline of Dmdmdxmice, but genome editing in the germline of humans is not practicable, and HDR does not occur readily in post-mitotic, adult skeletal muscle and heart. Therefore, an alternate approach was needed.
The Dmdmdxmutation is a point mutation in exon 23 of the Dmd gene that introduces a premature stop codon and results in a non-functional truncated protein the lacks the C-terminal dystroglycan binding domains. Exon 23 encodes amino acids that comprise several spectrin-like repeat motifs that comprise dystrophin’s large, central rod domain, and previous work had demonstrated that in-frame deletions that remove large chunks of these domains can produce truncated, but mostly functional dystrophin. Therefore, all three of the Science publication teams designed sgRNAs that would introduce double-stranded DNA breaks in the introns on either side of exon 23 and that, when healed by Cas9 NHEJ activity, would restore the dystrophin open reading frame and express a functional dystrophin isoform with exon 23 removed (see Figure 1).
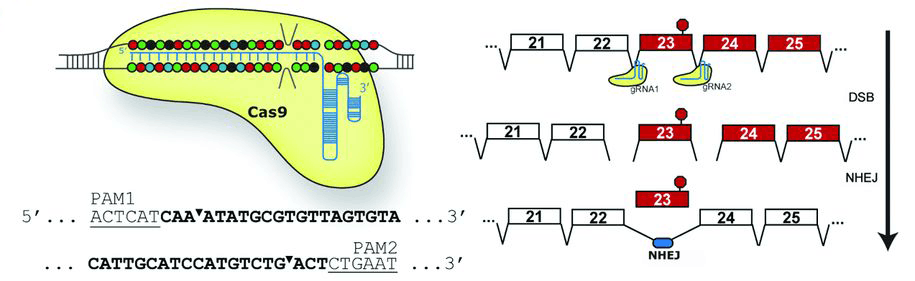
(Figure 1) A CRISPR/Cas9-mediated exon skipping approach to correct the Dmdmdxmutation. Paired guide RNAs (gRNA1) were designed that recognize PAM sequences (lower left) on either side of mouse Dmd exon 23 that carries the mdx point mutation (red circle; right image). Following Cas9-mediated double-stranded breaks (DSB) and non-homologous end joining (NHEJ), the aberrant exon 23 is removed, allowing an mRNA encoding a truncated, but functional dystrophin protein to be produced (image from Nelson, CE et al. 2016).
CRISPR/Cas9 genome editing in vivo restores dystrophin expression and improves muscle function
With their exon skipping strategies in place, the three investigative teams introduced both Cas9 and sgRNA expression constructs into young (P1-P18) Dmdmdxdystrophic mouse muscle via adeno-associated virus (AAV) delivery vectors. The viruses were delivered either locally via intramuscular (IM) injection into the tibialis anterior (leg muscle) of Dmdmdxmice or systemically via intraperitoneal (IP), intravenous (IV) or retro-orbital (RO) administration. In the UT Southwestern study (Long C et al. 2016) dystrophin-positive myofibers could be observed in the IM-injected muscle at 3 weeks post-treatment, and the percentage of dystrophin-positive fibers increase protein levels had increased 3 weeks later (7.7 +/- 3.1% positive @ 3 weeks; 25.5 +/- 2.9% @ 6 weeks). The Duke team (Nelson, CE et al. 2016) observed that approximately 67% of myofibers in IM-injected muscle showed dystrophin immunostaining at 8 weeks post-treatment, and dystrophin protein levels estimated from immunoblots of whole muscle lysates were at approximately 8% of wild-type levels (Figure 2A and B). Droplet digital PCR, Sanger sequencing and quantitative PCR were used by the various teams to confirm that exon 23 genome editing events had occurred or that mRNAs lacking exon 23 were transcribed. Further, histochemical staining of IM-treated muscle showed reductions in histopathological features that are hallmarks of dystrophic muscle, including fewer necrotic myofibers, reduced inflammatory cell infiltration, and few myofibers with centralized nuclei. Importantly, the Harvard/MIT team (Tabebordbar M et al. 2016) also verified that genomic editing events had occurred in muscle satellite cells - the normally quiescent muscle precursor cells that reside interstitially between myofibers and are activated to differentiate and form new muscle fibers following myofiber damage and degeneration. Dystrophin-positive myofibers, and dystrophin protein, also, were observed in systemically-treated mice, although the numbers and protein levels were somewhat reduced in muscle compared with the IM-treated mice. Importantly, however, dystrophin expression and dystrophin-positive cells were observed in cardiomyocytes. Because complications from cardiomyopathies frequently lead to death in DMD patients, rescuing dystrophic muscle phenotypes in heart is vital for any DMD therapy.
Intramuscular and systemic delivery of the Cas9 and sgRNAs also improved muscle function. In the various studies, forelimb grip strength, and various contractility assays to measure muscle force were improved in the CRISPR/Cas9-treated mice (Figure 2C).
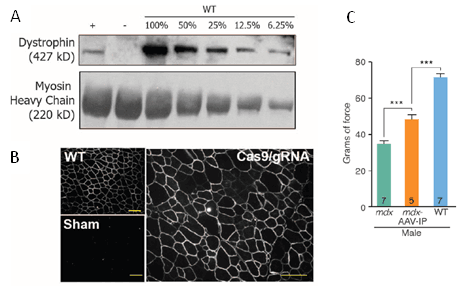
(Figure 2 A, B, & C) CRISPR/Cas9-genome editing rescues dystrophin expression and improves muscle function in vivo. Eight weeks after injecting Cas9/sgRNAs into dystrophic muscle, dystrophin protein expression (+ in A) was observed in whole muscle extract immunoblots and dystrophin-positive myofibers were seen by immunohistochemistry. Muscle function as assessed by forelimb grip strength was also improved by 4 weeks post-treatment systemically (via IP delivery of Cas9/sgRNA AAV vectors). (A and B from Nelson, CE et al. 2016; C from Long C et al. 2016).
Together, the three studies published in Science validate CRISPR/Cas9 genome editing as a potential new therapeutic approach for restoring in vivo dystrophin expression in DMD patients. Although overall dystrophin protein levels in the muscles and hearts of CRISPR/Cas9-treated Dmdmdx mice were only a fraction of the levels expressed in wild-type mice, previous studies have demonstrated that restoring dystrophin protein to levels as low as 4-15% can ameliorate DMD pathologies and restoring levels to 30% can suppress the DMD phenotype altogether. Further refinements to the Cas9/sgRNA delivery vectors may improve the efficiency with which editing within skeletal muscle and heart myofibers occurs, and support more robust restoration to muscle function.
We probably are still several years away from using CRISPR/Cas9 to treat DMD patients. One important issue that must be addressed with any use of the technique is the possibility for off-target genomic editing to introduce unwanted mutations at other sites in the genome. In the three Science studies, however, such off-target events at the PAM sites most similar to those used to delete Dmd exon 23 were either not detected, or occurred with very low frequency. If methods can be developed to better identify or prevent such events, CRISPR/Cas9 may hold the key to helping DMD patients enjoy longer and more active lives.