Human genomes vary quite a bit from individual to individual. These differences include single nucleotide changes, or “spelling mistakes” in the DNA sequence, but even more variation comes from structural variants, which include additions, deletions and rearrangements of large segments of DNA. A recent study used multiple advanced technologies to dive deeper than ever before to comprehensively characterize the structural variants present in three families, and what their functional consequences might be.
Genome sequencing has become much faster, more accurate and less expensive over the past decade. As a result, more and more human genomes are being sequenced, and our knowledge of what those sequences actually mean for function and disease is growing rapidly.
It has also become clear that the more we learn, the more we appreciate just how little we still know about human genomics. While a linear sequence of ATGCs looks tidy, our genomes are actually dynamic entities that harbor considerable differences between individuals — differences that can alter traits contributing to both normal function and disease.
The genetic difference between individuals contributes to our individuality. These differences include millions of single nucleotide variants — where, for example, one person may have an A, another may have a C at a given position. There are also hundreds of thousands of structural variants (SVs). SVs include segments of DNA that are inserted into or deleted from the genome, segments that are duplicated, and segments that are inverted. SVs are more difficult to identify than single nucleotide variants, and hence it has been unclear just how many SVs really exist in a given human genome.
Now a paper entitled, “Multi-platform discovery of haplotype-resolved structural variation in human genomes,” published in Nature Communications, delves deeper into individual genomic differences than ever before.
The work involved a large international team of researchers from the Human Genome Structural Variation Consortium (HGSVC), led by co-first authors Mark Chaisson, Ph.D., Ashley Sanders, Ph.D., and Xuefang Zhao, Ph.D.; co-senior authors Paul Flicek, Ph.D., Ken Chen, Ph.D., Mark Gerstein, Ph.D., Pui-Yan Kwok, M.D., Ph.D., Peter Lansdorp, M.D., Ph.D., Gabor Marth, DSc., Jonathan Sebat, Ph.D., Xinghua Shi, Ph.D., Ali Bashir, Ph.D., Kai Ye, Ph.D., Scott Devine, Ph.D., Michael Talkowski, Ph.D., Ryan Mills, Ph.D., and Tobias Marschall, Ph.D.; and co-corresponding senior authors Jan Korbel, Ph.D., Evan Eichler, Ph.D., and The Lee LabThe Lee Lab studies structural genomic variation in human biology, evolution and disease.Charles Lee, Ph.D., FACMG . They used a full suite of genomic technologies to extensively analyze the genomes of three family trios (parents and child). The technologies used include long-read, short-read, and strand-specific sequencing technologies, optical mapping and multiple computer algorithms for SV detection. The results present the most comprehensive catalog of SVs to date in the children’s genomes, including information on which set of parental chromosomes each SV was present on.
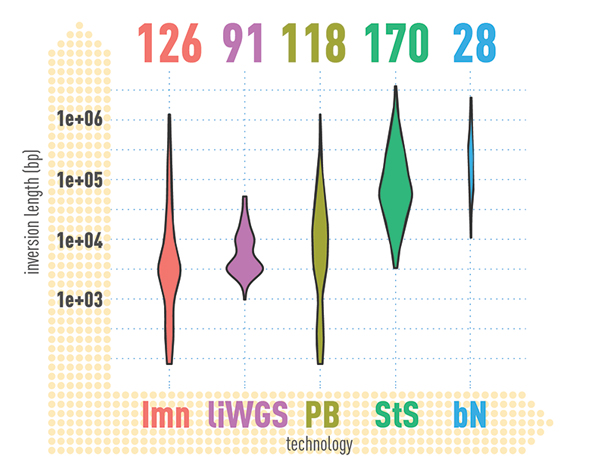
In summary, the researchers identified an average of 818,054 small insertions and deletions (genomic alterations that each affected less than 50 bases of DNA) and 27,622 SVs (genomic alterations that affected 50 bases or more of DNA) per genome. Remarkably, they also found an average of 156 inversions per genome, many of which intersected with genomic regions associated with genetic disease syndromes. The researchers found that more than 100,000 variants per individual are actually missed by routine sequencing technologies and commonly-used computer algorithms. For example, 83% of the insertions identified were missed by standard short-read-calling algorithms. In fact, the true numbers of SVs in a given human genome appears to be three- to seven-fold more than most studies typically identify.
Hence, SVs constitute a large amount of genetic variation not commonly captured by current genome sequencing technologies and analytical methods. This implies that the contribution of SVs to human disease has not yet been well-quantified and the expanded SV repertoire can help identify new genetic associations to diseases and improved diagnostic yields in future genetic tests.