Amyotrophic lateral sclerosis (ALS) involves progressive loss of nerve cells and neuromuscular function. About 5,000 people are diagnosed with ALS each year in the United States, and most people with ALS live for only two to five years after diagnosis. Despite extensive inquiry, the underlying cause(s) of most ALS cases remains unknown, and there is no cure. Recent research has revealed potential disease mechanisms, however, as well as possible therapeutic targets.
An important development in ALS research has been investigations into TAR DNA-binding protein 43 (TDP-43), which plays a vital role in regulating the levels and proper splicing of pre-messenger RNAs (pre-mRNAs) from more than 1,500 genes. Nearly two decades ago, scientists found that a group of neurological disorders are associated with the mislocalization of TDP-43 outside the nucleus and aggregation in the cellular cytoplasm. Strikingly, mislocalized TDP-43 is found in more than 95% of affected neurons in amyotrophic lateral sclerosis (ALS) cases, and at least half in frontotemporal dementia (FTD). More recently, research has revealed that normal nuclear TDP-43 function is particularly essential for proper maturation of the pre-mRNA encoding stathmin-2, coded for by the STMN2 gene, a protein necessary for the outgrowth of neurons’ axons during development and regeneration following any injury. TDP-43 suppression in human cells results in a shortened, non-functional stathmin-2 mRNA and a loss of stathmin-2 protein.
But what are the consequences? An early hypothesis was that stathmin-2 regulates the rapid growth and shrinkage of microtubules in nerve cells. The full picture of TDP-43/stathmin-2 dysfunction was difficult to acquire, however, in part because, unlike humans, TDP-43 mislocation does not drive stathmin-2 loss in mice. A research team led by Jackson Laboratory (JAX) Vice President, Rare Disease Translational Center Cathleen Lutz, Ph.D.; Professor, Cellular and Molecular Medicine Don Cleveland, Ph.D., of the University of California at San Diego; and Associate Professor of Neurology Clotilde Lagier-Tourenne, Ph.D., of Harvard Medical School and the Broad Institute of MIT and Harvard, therefore took a different approach. They worked to directly disrupt stathmin-2 function in mice to investigate the effects on motor neuron structure and function. In “Stathmin-2 loss leads to neurofilament-dependent axonal collapse driving motor and sensory denervation,” published in Nature Neuroscience, the researchers show how stathmin-2 contributes to the establishment and maintenance of large-diameter axons needed for motor function, and they propose a new therapeutic approach to ALS and other neurodegenerative diseases.
The maintenance of motor function
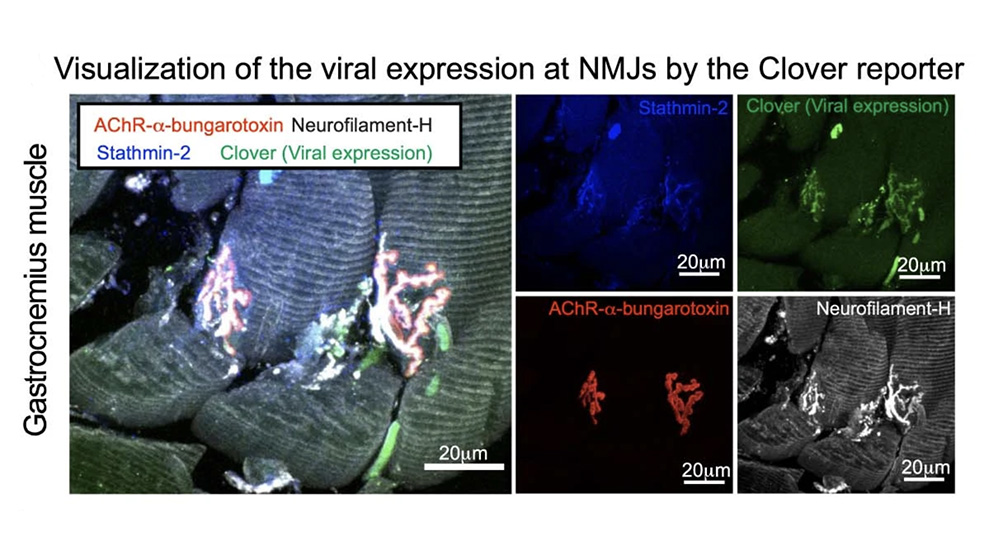
The research team used multiple methods to suppress or eliminate stathmin-2 in mice. In adult wild-type (unaltered) mice, they used antisense oligonucleotides (ASOs) that target the stathmin-2 mRNA to suppress its function and protein production in the nervous system for two to eight weeks. Although the mice had no outward appearance of disease, the nerve conduction velocity within the sciatic nerve was significantly reduced and, notably, the integrity of neuromuscular synapses, which drive muscle function, was compromised. The researchers then engineered a system to chronically suppress stathmin-2 mRNA and protein production using short hairpin RNAs (shRNAs) delivered to the spinal cord through viral vectors. Over the course of eight months, the mice exhibited impaired motor performance and the disruption of more than half of the neuromuscular junctions of lumbar motor neurons, with 43% fully denervated. There was also a reduction in the conduction velocity of the sciatic nerve that was similar to the reductions reported in patients with ALS.
Investigating the nerve cells themselves following chronic stathmin-2 suppression, the researchers found substantial changes to the axons, the long projections of nerve cells that conduct electrical impulses through the body. The axons shrank, with some types of axons losing as much as 50% of their cross-sectional area. Also, the myelin sheath, the insulating layer that wraps around the axon and is essential for proper nerve impulse conduction, became delaminated, with tearing in the outer layers. The internal structure of the axons themselves appeared collapsed, but instead of microtubule disruption, the team found changes to neurofilaments, another key structural component of neurons. The neurofilament organization became abnormal, with highly compacted compartments. Importantly, the findings mirror those from the evaluation of postmortem human samples from ALS patients, which revealed similar axonal collapse and myelin tearing.
But what happens if stathmin-2 is absent from the start? Inactivating the gene from birth yielded low survival rates shortly after birth in one mouse strain, indicating that stathmin-2 contributes to early development. Crossing the mice with another strain improved survival, however, indicating the background genetics also play an important role. In any event, the surviving mice of both strains that lacked stathmin-2 from birth exhibited traits similar to the mice with chronic stathmin-2 suppression: significantly impaired motor performance by three months of age, progressive denervation of hindlimb muscles and reduced conduction velocity in the sciatic nerve over the course of a year, reduced axon diameters, and disrupted neurofilaments.
Potential cause, potential therapy
The paper’s findings indicate that stathmin-2 is needed for maintaining motor neuron axons and both motor and sensory neuron synapses. Sustained suppression of stathmin-2 in mouse models disrupts synapses and triggers a degenerative process similar to that observed in the early stages of ALS. Therefore, loss of stathmin-2 function is strongly implicated as an underlying cause of ALS initiation and progression. While other factors likely contribute to the full picture of ALS disease pathology, the results suggest that restoring stathmin-2 is a promising therapeutic approach for ALS and other TDP-43-dependent neurodegenerative diseases.